JSMC Biochemistry and Molecular Research
-
Letter to EditorThe Unique Molecular Structure of Human T-Cell Leukemia Virus Type 1 ProteaseMohsen Karbalaei1 and Masoud Keikha2*1Department of Microbiology and Virology, Jiroft University of Medical Sciences, Iran
2Department of Microbiology and Virology, Mashhad University of Medical Sciences, Iran*Corresponding author: Masoud Keikha, Department of Microbiology and Virology, Mashhad University of Medical Sciences, Mashhad, Iran, Tel: 09386836425; Email: masoud.keykha90@gmail.comSubmitted: 23 April 2019; Accepted: 29 April 2019; Published: 30 April 2019 -
Human T-cell leukemia virus type 1 (HTLV-1) is retrovirus type C which causative agent of Human T-cell leukemia virus type 1 associated myelopathy/tropical spastic paraparesis (HAM/ TSP), Adult T-cell leukemia/lymphoma (ATL or ATLL), uveitis, arthritis, alveolitis/bronchiectasis, dermatitis, and lymphadenitis [1]. There are approximately 15-20 million people were infected by HTLV-1 that majority of them where lives in HTLV-1 endemic area including North America, Central Africa, Caribbean islands, Japan, Australia and Iran (particularly Khorasan province) [1-2]. Although more than 90% of the HTLV-1 infected individuals are remains as asymptomatic carrier but 2-6% of HTLV-1infected individuals are progress to ATLL, as well as 2-3% develop to HAM/TSP [3].HTLV-1 is transmitted by unsafe sexually contact, injection, blood transfusion and breastfeeding throughout population [2,3]. There is no selective treatment option against HTLV-1; combination of AZT plus INF-α is only available therapeutic option of HTLV-1 infection which has not efficient completely [4]. It is suggested that molecular targeting of HTLV-1 has considered as the best strategy for appropriate treatment; of which, the Protease enzyme is the suitable option because of its play key role in process of HTLV-1 polyprotein and has critical role in HTLV-1 pathogenesis [5]. Therefore, molecular targeting of HTLV-1 protease has one of the interest option for development of selective antiviral compounds against HTLV-1 [3,5]. But it’s necessary to making a survive about structural conformation and sequence dissimilarities between different strains of HTLV-1 to design therapeutic option based on the conserved domains of HTLV-1 Protease which inhibits all strains of HTLV- 1. Therefore, the of this study was comparative analysis and molecular structure study of the HTLV-1 Protease molecules for determination of dissimilarities and conserved domains of HTLV- 1 protease for construct of selective HTLV-1 protease inhibitors.Initially, total of 100 HTLV-1 protease amino acid sequences of different regions were obtained from NCBI (https://www. ncbi.nlm.nih.gov/protein); then multiple alignment and the phylogenic tree was constructed using the neighbor-joining method with Kimura‐2‐parameter (K2P) distance for evaluation of genetic diverge among HTLV-1 strains. According to our results, multiple alignment study (via ClustalW) was showed that highest similarity in the sequence of HTLV-1 Protease among different HTLV-1 strains (100-98.93% similarity); also, the phylogenic tree was approved the majority of similarity in target gene sequences (Figure 1).
-
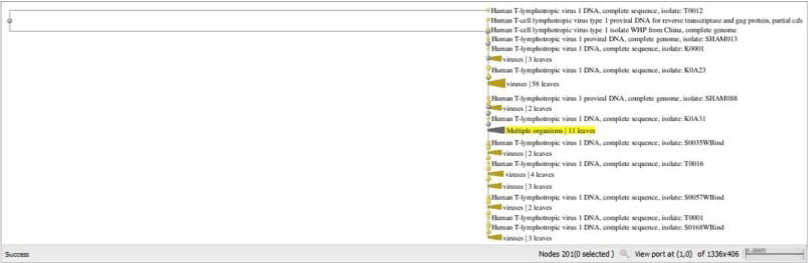 Figure 1:The Phylogenetic tree of the one hundred sequences of HTLV-1 protease using the neighbor joining method. View Figure
Figure 1:The Phylogenetic tree of the one hundred sequences of HTLV-1 protease using the neighbor joining method. View Figure
Subsequently, the crystal structure of HTLV-1 protease was obtained from PDB (3WSJ) for determination of active site (Figure 2). Then, entirely of the crystal structures of HTLV-1 Protease were obtained from PDB (https://www.rcsb.org/) and multiple aligned in order to discovery of dissimilarity of the sequences (Figure 3). We confirmed that amino acid sequence of the active site of HTLV-1 is nearly conserved complete among different HTLV-1 strains.-
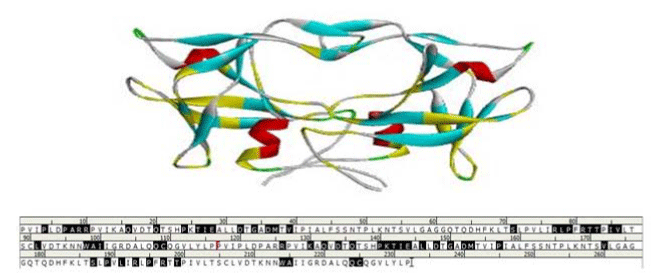 Figure 2:The active site of HTLV-1 Protease was highlighted in the crystal structure and amino acid sequence. View Figure
Figure 2:The active site of HTLV-1 Protease was highlighted in the crystal structure and amino acid sequence. View Figure
-
 Figure 3:Multiple alignment of HTLV-1 Protease sequences. View Figure
Figure 3:Multiple alignment of HTLV-1 Protease sequences. View Figure
Finally, about 100 crystal structure of HTLV-1 Protease were superimposed for 3D dimensional and conformation analysis of the HTLV-1 Protease molecule which showed that a strong similarity among HTLV-1 Protease structures and confirmed our previous results about homology of HTLV-1 Protease in different strains (Figure 4).-
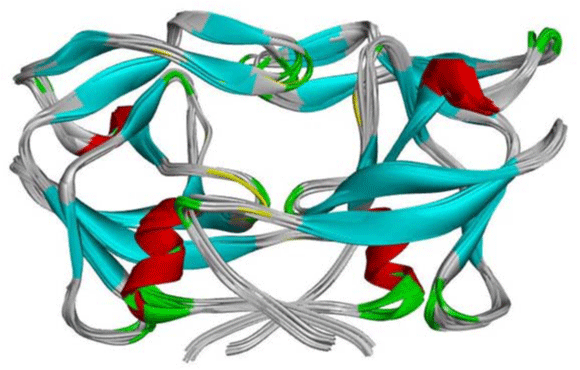 Figure 4:The superimposed of one hundred crystal structure of HTLV-1 Protease. View Figure
Figure 4:The superimposed of one hundred crystal structure of HTLV-1 Protease. View Figure
Overall, HTLV-1 protease is aspartic protease which responsible for maturation hydrolysis of HTLV-1 polyproteins throughout active site includes two aspartic acid residues (Asp32, 36), it’s expressed in homodimeric form with each chain consisting of 125 residues that is highly specific to its substrate [6]. In recently years, the HTLV-1 Protease is known as the suitable molecular targets for efficiently treatment of HTLV-1 infection. According to us survive, the active site of the HTLV-1 Protease is conserved among different strains of HTLV-1 and can be used as target for development of HTLV-1 Protease inhibitors. -
-
- Boxus M, Willems L. Mechanisms of HTLV-1 persistence and transformation. Br J Cancer. 2009; 101: 1497-1501.
- Bertazzoni U, Ciminale V, Romanelli MG. Molecular Pathology of HTLV-1. Front Microbiol. 2018; 9: 3069.
- Soltani A, Hashemy SI, Avval FZ, Soleimani A, Rafatpanah H, Rezaee SA, et al. Molecular targeting for treatment of human T-lymphotropic virus type 1 infection. Biomed Pharmacother. 2019; 109: 770-778.
- Macchi B, Balestrieri E, Frezza C, Grelli S, Valletta E, Marçais A, et al. Quantification of HTLV-1 reverse transcriptase activity in ATL patients treated with zidovudine and interferon-α. Blood Adv. 2017; 1: 748-752.
- Selvaraj C, Singh P, Singh SK. Molecular modeling studies and comparative analysis on structurally similar HTLV and HIV protease using HIV-PR inhibitors. J Recept Signal Transduct. 2014; 34: 361-371.
- Kuhnert M, Steuber H, Diederich WE. Structural basis for HTLV-1 protease inhibition by the HIV-1 protease inhibitor indinavir. J Med Chem. 2014; 57: 6266-6272.
Search Articles
-
Short Communication
Why Multi-Drug Antiviral Therapy is Needed for COVID-19
Sibasish Dolai1 , Sabine Hazan2 , Christelle Pagonis1 , Sabrina Liu3 , Thomas J Borody1* and Robert R Clancy1 | 2024-04-11- Review Article
Elja: An R Package to Perform Environment-Wide Association Studies (Ewas / Envwas) Analysis
Marwan El Homsi1*and Isabella Annesi-Maesano1,2 | 2024-03-25- Case Report
Diagnosis of a Neck Mass in the Emergency Department
Schmidt S¹,²*, Lorenz KJ¹, Matthias C², Diekmeyer B2 and Müller G?,? | 2024-03-14- Case Study
The Impact of Neuropsychiatric Symptoms of Alzheimer’s Disease on Family Caregiver’s Distress
ATomasello L1,2*, Ranno1 , Raffaele2 , Laganà3 , Pitrone3 and Alibrandi4 | 2024-02-28- Mini Review
Metabiotics as Potential Therapeutic Agent in Mucosal Immunity
Md Zeyaullah 1*, Irfan Ahmad2 , Faruque Ahmad3 , S Rehan Ahmad4 , Razi Ahmad5 , Abdullah M AlShahrani1 , Mohammad Suhail Khan6 , Mohammad Shane Alam7*, Adam Dawria6 , Atiq Hassan1 , Khursheed Muzammil6 , Haroon Ali6 and Zaki H Hakami7 |News Feeds

Seth J. Worley, MD, FHRS, FACC
Director, Interventional Implant Program
Biography Paper Presentations
MedStar Heart & Vascular Institute,
Washington, DC, USACollaborations
Indexing







 © Copyright - JSM Central
© Copyright - JSM Central This work is licensed under a Creative Commons Attribution 4.0 International License.
This work is licensed under a Creative Commons Attribution 4.0 International License.
- Review Article