High-Dose Statin Associated with Rhabdomyolysis, Acute Kidney Injury, Cholestatic Liver Injury, and Thrombocytopenia
Introduction: Statins are the drugs of choice to reduce cholesterol and the incidence of cardiovascular events. Although rare, the side effects of these drugs may be severe (especially when given in the high doses recommended by the cardiologists), including: muscle damage, renal and liver injury and compromised function, and polyneuropathy.
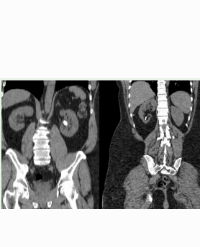
Case Report: We report a case of statin-induced rhabdomyolysis, acute kidney and liver failure and thrombocytopenia that developed in a 76-year-old man, who was referred to our department because of severe generalized myalgia and muscle weakness, extreme fatigue, loss of appetite, dark brown urine. Following an acute myocardial infarction 8 months previously he was put on atorvastatin 80 mg once daily. Laboratory evaluation at presentation revealed much increased levels of muscle enzymes, aminotransferases, total and conjugated bilirubin, and nitrogenous waste products, and low platelets. A diagnosis of acute renal and liver failure secondary to the long-term intensive statin therapy was made. Atorvastatin was discontinued and forced alkaline diuresis was started. After five days of oliguria and slight but persistent increase in creatinine levels dialysis was initiated, but discontinued after 4 sessions, once urine output increased. At discharge the patient’s serum creatine kinase level was in the normal range, creatinine was significantly decreased the thrombocyte count was better, aminotransferase were much lower but not completely normalized, but the bilirubin remained at the same level. The patient was discharged and instructed to avoid any potentially nephrotoxic and hepatotoxic drugs until next outpatient evaluation.
Conclusions: Our case report is meant to raise concerns about prescribing high dose statins. Unfortunately the prescribing cardiologists may be insufficiently aware of the potential for severe adverse effects as these come to the attention of clinicians from different specialities, especially nephrologists.
Dorin Dragos1,2, Diana Pruteanu2 and Rodica Constantin2