Short Communication | Volume 5 - Issue 1 | Article DOI :
Download PDF
Stylianos Velonis¹, Panagiotis Skaltsounis² and Anastasia E Konstantinidou²*
¹Orthopedic Clinic, Metropolitan General Hospital, Athens, Greece
²Department of Pathology, Unit of Perinatal Pathology, Medical School, National Kapodistrian University of Athens, Greece
Corresponding Author:
Anastasia E Konstantinidou, Departmentof Pathology, Unit of Perinatal Pathology,Medical School, National Kapodistrian University of Athens, Greece,Tel: +30 210 7462159;Fax : +30 210 7462179;Email: ankon@med.uoa.gr
Keywords
Cilia; Ciliopathy; Fetus, Skeletal dysplasia, Perinatal autopsy, Phenotype; Postmortem
Abstract
Skeletal ciliopathies are inherited genetic skeletal dysplasias which result from defects in the biosynthesis and/or function of primary cilia. The predominant skeletal phenotype includes a narrow trunk with markedly or variably short ribs, and short limbs with or without polydactyly. Skeletal ciliopathies are commonly associated with particular extraskeletal malformations, variably including hindbrain malformations, corpus callosum agenesis, retinal, cardiovascular, gastrointestinal and genital abnormalities, and a particular spectrum of hepatorenal changes. The most severe among skeletal ciliopathies become manifest during fetal life and can be prenatally detected by ultrasound, leading to termination of pregnancy due to poor prognosis. The fetal skeletal ciliopathies are inherited in an autosomal recessive way, thus bearing a 25% recurrence risk in subsequent pregnancies, in contrast to the majority of fetal skeletal dysplasias which represent de novo mutations of autosomal dominant conditions with a very low recurrence risk. The phenotypic diagnosis is mainly based on the radiographic features in association with the external, visceral and histological findings at postmortem examination. In this article we review the fetal phenotype of the skeletal ciliopathies and provide a review of the ciliary mutations that have been associated with each entity.
Introduction to the ciliary function
The term “skeletal ciliopathies” refers to inherited genetic skeletal dysplasias which result from cilia malfunction affecting skeletal development. Cilia are hair-like organelles projecting from the surface of cells.
They are divided into two main subgroups, motile and non-motile or primary cilia. Motile cilia, distinguished by their ability to beat rhythmically, occur in bundles of hundreds, and are topographically restricted to the respiratory tract, brain ventricles, and reproductive tract, where they organize fluid flow across the cell surface, move extracellular fluid and, by flushing away bacteria and other debris, provide the mucociliary clearance.
Motile cilia are also found in a primitive embryonic structure in mammals, the embryonic node, where they beat and produce a leftward fluid flow so that the embryo knows left from right and can establish the normal left-right asymmetry, e.g. place the heart to the left and the liver to the right.
In mice, when motile ciliary function is impaired due to mutant cilia that fail to beat, as a result the leftward nodal flow is not established and mouse embryos may develop laterality defects, with their internal organs on the wrong side [1]. In humans, mutant motile cilia also lead to Primary Ciliary Dyskinesia (including Kartagener syndrome), a cystic fibrosis-like disease presenting with bronchiectasis, infertility, and laterality defects [2].
Primary cilia are non-motile single structures that can be found on nearly every cell throughout the organism, projecting like an antenna from the cell surface. They have chemosensory, osmosensory and phototransduction functions.
The eye, cerebellum, liver, kidney, and bone are vulnerable to the ciliary dysfunction [3]. The ciliary proteins are produced in the cell and selectively transported into the cilium via the intraciliary transport, more commonly called intraflagellar transport (IFT), as cilia are structures very similar to flagellae. Anterograde IFT from the ciliary base to the tip is facilitated by the kinesin motor proteins belonging to the IFT complex B. The IFT complex-B provides the power to move the ciliary protein cargo from the cytoplasm to the ciliary tip, while cargo may be unloaded along the cilium and at the tip [4].
From the ciliary tip, molecules are conveyed back to the cytoplasm at the ciliary base by retrograde transport using the IFT complex-A, which is catalyzed by the dynein 2 complex molecular motor, which provides the power for ciliary assembly [5,6]. The IFT complex-B consists of nine core components and several peripheral subunits, while the IFT complex-A consists of six primary components and other ancillary proteins (reviewed by Zhang et al, 2018) [7].
An expanding group of syndromic genetic conditions with a broad range of phenotypes are linked to malfunction of primary cilia [8,9]. These ciliopathies include among others the Polycystic Kidney Diseases, Meckel-Gruber and Joubert syndromes, Bardet-Biedl syndrome, Nephronophthisis, and the skeletal ciliopathies. These comprise a group of genetic skeletal dysplasias that share common skeletal changes and are often associated with hepatorenal, cerebellar, genital, and retinal malformations, singly or in combination [9-11].
Types of skeletal ciliopathies
The skeletal ciliopathies known to date include the perinatal lethal Short-Rib polydactyly syndromes (SRPs) and the overlapping but less severe, compatible with life phenotypes of Asphyxiating Thoracic Dystrophy Jeune (ATD Jeune), Chondroectodermal dysplasia or Ellis-van Creveld (EvC) syndrome, Orofaciodigital syndrome type IV (OFD IV), Mainzer-Saldino syndrome (MZSD), Cranioectodermal Dysplasia (CED), as well as Weyers Acrodental Dysostosis (WAD), a condition allelic to EvC which does not affect the thorax. With the exception of the autosomal-dominant WAD, all the remaining known to date skeletal ciliopathies, lethal and nonlethal, are autosomal recessive diseases and may present at the fetal and perinatal age with a variable degree of severity. The various types of SRPs, and severe forms of EvC and ATD Jeune are included among the more commonly encountered and diagnosed fetal skeletal dysplasias, prenatally detected by ultrasound as early as the first trimester of gestation, [12] and commonly leading to termination of pregnancy due to poor prognosis, based on the degree of thoracic constriction and limb shortening [13-15].
The various types of short-rib-with-or-without-polydactyly syndromes described below are based on a phenotypic classification, while all the skeletal ciliopathies featuring short ribs and thoracic constriction are grouped in MIM under the label of Short-Rib Thoracic Dysplasias (SRTD 1 to 20), according to the mutations in ciliary genes identified so far.
Phenotypic features
The predominant skeletal phenotypic features in most skeletal ciliopathies include a narrow trunk with markedly or variably short ribs, and short limbs with or without polydactyly. Skeletal ciliopathies are commonly associated with particular extraskeletal malformations, such as Dandy-Walker malformation, corpus callosum agenesis, cardiovascular, gastrointestinal and genital abnormalities, and a specific spectrum of hepatorenal changes. These associations appear to characterize many diseases that fall into the large and expanding group of ciliopathies [9,16,17].
Short-Rib with or without Polydactyly syndromes (SRPs)
There are at least 4 phenotypically distinct types: type I (SaldinoNoonan syndrome, MIM #613091), type II (Majewski syndrome, MIM #263520), type III (Verma-Naumoff syndrome, MIM #613091) and type IV (Beemer-Langer syndrome, MIM %269860). A further type V of SRP, similar to type III with acromesomelic hypomineralization and camptomelia has also been identified (MIM #614091).
Types I and III have been conventionally considered to represent a phenotypic spectrum of the same entity, as types II and IV. Despite the similarities, however, each subtype presents specific phenotypic features.
Overlapping characteristics can be seen between all 5 types and several cases are described that cannot fit in a particular type, this phenotypic variability being a characteristic of ciliary diseases. Severe thoracic constriction is a hallmark in all SRP types, which are invariably lethal in the early neonatal period due to the consequent pulmonary hypoplasia.
The various phenotypic forms of SRPs differ by visceral malformation and metaphyseal appearance on the X-ray.
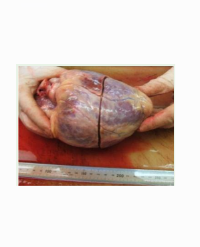
External inspection in SRPs reveals a long narrow trunk, short limbs, and polydactyly (Figures 1 and 2). Polydactyly is postaxial in SRP types I, III and V, preaxial or combined preaxial and postaxial in SRP type II, rare in SRP type IV [14,15]. The combination of these findings contributes to an early sonographic prenatal diagnosis, achieved as early as the 12th week of gestation [12]. SRP-I in addition may present with fetal hydrops. The typical hallmark of SRP-I is extreme micromelia, sometimes resulting in ‘flipper-like’ limbs.

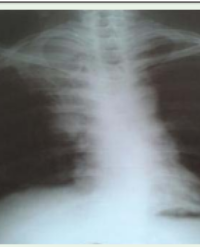
Figure 1: SRP-I (Saldino-Noonan syndrome). A: Small chest, distended abdomen, very short limbs, postaxial polydactyly. B: X-ray shows extremely short and horizontal ribs, shortened and rounded iliac bones with acetabular spikes, very short tubular bones with spiked metaphyseal ends (inset).

Figure 2: SRP-II (Majewski syndrome). A: Flat nose, narrow trunk, extreme polydactyly in hands and feet. B: X-ray shows V-shaped middle metacarpals (arrow) and the tibia shorter than the fibula. C: Micropenis.
X-ray principal features (Figures 1 and 2) include short horizontal ribs, short tubular bones and small iliac bones with trident acetabulae (horizontal acetabular roofs with spur-like projections at the lower margins of the sciatic notches). Metaphyseal spurs characterize SRP-I and are more prominent in SRP-III, while the metaphyseal ends are smooth in types II and IV [18]. The tibia is shorter than the fibula or short and oval-shaped in SRP-II (Majewski) [14,18]. V-shaped fusion of metacarpals can also be seen in SRP-II (Figure 2B). Handlebar clavicles have been observed in cases with radiological features consistent with SRP-III.
Visceral pathology includes cystic kidneys, increased portal mesenchyma, persisting ductal plate or ductal plate malformation of the liver, cardiac and gastrointestinal defects, as well as genital defects ranging from micropenis to aplasia of the external genitalia.
Chondroectodermal dysplasia /Ellis-van Creveld (EvC) syndrome (MIM #225500) and ATD Jeune (MIM #208500) also belong to the group of Short-Rib Thoracic Dysplasias. These two may be phenotypically indistinguishable in the fetus. Compared to SRPs, the phenotype is less severe. Polydactyly is an inconstant feature of ATD-Jeune and, when present, usually also affects the feet. In contrast, postaxial polydactyly of the hands is a constant feature in EvC, but the feet are uncommonly affected [11]. In cases of ATD Jeune with polydactyly, differentiation from EvC may not be possible on radiologic grounds alone. EvC may also be indistinguishable from SRP-II in the fetal period. Surviving EvC patients develop in childhood the typical disorders of ectodermal dysplasia, i.e. hypoplastic nails, thin hair and abnormal teeth. Poor nail formation may be the only sign of ectodermal disorder in the fetus, thus favoring the diagnosis of EvC versus ATD Jeune or SRP-II.
On X-ray the thorax is narrow, less severely affected than in SRPs, the tubular bones are short with smooth metaphyseal ends, and the ilia are shortened with trident acetabulae. V-shaped metacarpals may be a feature of EvC, similarly to SRP-II/Majewski [18]. Handlebar clavicles have been reported in ATD-Jeune [7].
The main visceral abnormality in ATD Jeune is renal, whereas it is cardiac in EvC (Table 1).
Table 1: Ciliary genes associated with the fetal skeletal ciliopathies.

Prognosis
Prognosis of the fetal skeletal ciliopathies is mainly based on the degree of thoracic constriction and the extent of limb shortening. The most severe thoracic constriction is usually seen in SRPs and JeuneATD. SRPs are considered to be invariably lethal in the perinatal period due to cardiorespiratory failure [10].
Ellis-van Creveld and ATD-Jeune may be compatible with life. ATD-Jeune shows over 50% perinatal mortality, especially in cases with dynein-2 complex mutations [19,20]. EvC may be compatible with life, but frequently shows severe heart defects. Mainzer-Saldino syndrome (MZSDS) and Cranioectodermal Dysplasia (CED) become manifest and are diagnosed after birth, in infancy or childhood, having a milder thoracic constriction but a higher rate of renal, retinal and liver disease that may lead to severe morbidity or mortality in childhood [17].
Molecular Genetics
The short-rib skeletal ciliopathies are grouped in MIM under the label “Short Rib Thoracic Dysplasias” (SRTD 1-20) according to the underlying ciliary mutation identified so far.
To date mutations in 23 genes have been found in cases of lethal and/or non-lethal skeletal ciliopathies [21]. Allelic heterogeneity in a number of the genes results in variable phenotypes along a wide spectrum of skeletal severity and extraskeletal manifestations. Table 1 shows the molecular basis of each type in the group of fetal skeletal ciliopathies, overall correlated with 15 ciliary genes so far [7,11,22-24]. These pathogenic mutations are homozygous or compound heterozygous, accounting for the autosomal recessive mode of inheritance in this group. The molecular basis of SRP-IV has only recently been revealed [7,24]. Common mutations shared between SRP-I / SRP-III and SRP-II / SRP-IV demonstrate that these syndromes with overlapping phenotypes are indeed allelic. More than one genes appear to account for each entity, ATD-Jeune being the most heterogeneous genetic disorder in this group [19]. Most of the depicted genes encode proteins of the IFT complex-A and the associated motor complex, cytoplasmic dynein-2. By contrast, mutations in only a subset of the genes encoding IFT complex-B members have been recently identified. In addition, mutations encoding for basal body, centrosomal and centriolar proteins have also been reported (gene mutations reviewed in Zhang et al. 2018) [7]. Mutations in several of these genes are rare causes of skeletal ciliopathies, each having been observed in only one or a few families.
In conclusion, skeletal ciliopathies constitute a group of genetically and phenotypically related skeletal dysplasias, yet presenting significant phenotypic variability and overlapping, on the grounds of genetic heterogeneity and involvement of multiple genes responsible of the biosynthesis or function of primary cilia. The severe skeletal phenotype with a narrow trunk, short ribs and short limbs with or without polydactyly, allows the majority of skeletal ciliopathies to become manifest during the fetal period, leading to termination of pregnancy or perinatal death. The fetal skeletal ciliopathies are inherited in an autosomal recessive way, thus bearing a 25% recurrence risk in subsequent pregnancies, in contrast to the majority of fetal skeletal dysplasias which represent de novo mutations of autosomal dominant conditions with a very low recurrence risk. Therefore, the identification of this group of disorders is essential, and is feasible on the basis of the postmortem skeletal radiographic, visceral and histological phenotypic findings. Targeted molecular confirmation will enable genetic counseling and prenatal diagnosis in future pregnancies.
Aknowledgments
Prof. A. Konstantinidou obtains funding for research and diagnosis in Fetal and Perinatal Pathology by “REA” Maternity Clinic, Athens, Greece.
References
1. Lowe LA, Supp DM, Sampath K, Yokoyama T, Wright CV, Potter SS, et al. Conserved left-right asymmetry of nodal expression and alterations in murine situs inversus. Nature. 1996; 381: 158-161.
2. Fliegauf M, Benzing T, Omran H. When cilia go bad: Cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007; 8: 880-93.
3. Anderson CT, Castillo AB, Brugmann SA, Helms JA, Jacobs CR, Stearns T. Primary cilia: Cellular sensors for the skeleton. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2008; 291: 1074-1078.
4. Scholey J M. Intraflagellar transport motors in cilia: Moving along the cell’s antenna. J Cell Biol. 2008; 180: 23-29.
5. Ishikawa H and Marshall WF. Ciliogenesis: Building the cell’s antenna. Nature Rev Mol Cell Biol. 2011; 12: 222-234.
6. Taschner M, Bhogaraju S, Lorentzen E. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation. 2012; 83: S12-S22.
7. Zhang W, Taylor SP, Ennis HA, Forlenza KN, Duran I, Li B, et al. Expanding the genetic architecture and phenotypic spectrumin the skeletal ciliopathies. Human Mutation. 2018; 39: 152-166.
8. Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006; 7: 125-148.
9. Baker K, Beales PL. Making sense of cilia in disease: The human ciliopathies. Am J Med Genet Part C Semin Med Genet. 2009; 151C: 281-295.
10. Huber C and Cormier-Daire V. Ciliary disorder of the skeleton. Am J Med Genet C Semin Med Genet. 2012; 160C: 165-74.
11. Schmidt M. Clinical genetics and pathobiology of ciliary chondrodysplasias. J Pediatr Genet. 2014; 3: 46-94.
12. Eleftheriades M, Iavazzo C, Manolakos E, Hassiakos D, Petersen M, Konstantinidou A. Recurrent short rib polydactyly syndrome. J Obstet Gynaecol. 2013; 33: 14-16.
13. Konstantinidou AE, Agrogiannis G, Sifakis S, Karantanas A, Harakoglou V, Kaminopetros P, et al. Genetic skeletal disorders of the fetus and infant: Pathological and molecular findings in a series of 41 cases. Birth Defects Res (Part A). 2009; 85: 811-821.
14. Konstantinidou AE. Skeletal Dysplasias of the Human Fetus: Postmortem Diagnosis. In: Prenatal Diagnosis - Morphology Scan and Invasive Methods. 1st ed. Choy RKW, Leung TY, (Editors), InTech 2012; pp.33-58.
15. Wyatt-Ashmead J, Konstantinidou AE, Offiah A. Skeletal dysplasias. In: The Pediatric and Perinatal Autopsy Manual. 1st ed. Cohen MC, Scheimberg I (Editors) Cambridge University Press 2014; p.235-261.
16. Waters AM and Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol. 2011; 26: 1039-1056.
17. Konstantinidou AE, Fryssira H, Sifakis S, Karadimas C, Kaminopetros P, Agrogiannis G, et al. Cranioectodermal Dysplasia: A Probable Ciliopathy. Am J Med Genet. 2009; 149A: 2206-2211.
18. Hall CM, Offiah A, Forzano F, Lituania M, Fink M and Krakow D. Fetal and Perinatal Skeletal Dysplasias, an atlas of multimodality imaging. Radcliffe Publishing Ltd, London, 2012; pp. 167-205.
19. Baujat G, Huber C, ElHokayem J, Caumes R, Claire DNT, David A, et al. Asphyxiating thoracic dysplasia: Clinical and molecular review of 39 families. J Med Genet. 2013; 50: 91-98.
20. Tuysuz B, Baris S, Aksoy F, Madazli R, Ungur S, Sever L. Clinical variability of asphyxiating thoracic dystrophy (Jeune) syndrome: Evaluation and classification of 13 patients. Am J Med Genet Part A. 2009; 149a: 1727-1733.
21. Mitchison HM and Valente EM. Motile and non-motile cilia in human pathology: From function to phenotypes. J Pathol. 2017; 241; 564-564.
22. Hoffer J, Fryssira H, Konstantinidou A, Ropers H-H, Tzschach, A. Novel WDR35 mutations in patients with cranioectodermal dysplasia. Clin Genet. 2013; 83: 92-95.
23. Huber C, Wu S, Kim AS, Sigaudy S, Sarukhanov A, Serre V, et al. WDR34 mutations that cause short-rib polydactyly syndrome type III/severe asphyxiating thoracic dysplasia reveal a role for the NF-κB pathway in cilia. Am J Hum Genet. 2013; 93: 926-31.
24. Silveira KC, Moreno CA, Cavalcanti DP. Beemer-Langer syndrome is a ciliopathy due to biallelic mutations in IFT122. Am J Med Genet A. 2017; 173: 1186-1189.
Citation
Velonis S, Skaltsounis P and Konstantinidou AE. Skeletal Ciliopathies: The Fetal Phenotype and Genetic Correlates. SM J Clin Med. 2019; 5(1): 1039s.