Research Article | Volume 4 - Issue 1 | Article DOI :
Download PDF
Lingzhen Cao* and Xiangxia Bu
College of Life Science, Jiangxi Normal University, China
Corresponding Author:
Lingzhen Cao, College of Life Science, Jiangxi Normal University, Ziyang District #99, Nanchang, CN 330022, China, Tel: 86-079188120391, E-mail: clzclz1011@163.com
Keywords
16S rRNA; Gut microbes; Diversity Analysis; Dragonfly
Abstract
Dragonflies are natural enemies for a host of insects, which mainly feed on mosquitoes and other insect pests. Bacterial communities in the dragonfly gut impact on host survival, ecology and evolution; however intestinal microbiota diversity in these insects is not well understood. In this study, we used the Illumina Miseq PE300 platform to characterize intestinal microbiota communities in four dragonfly species(Pantala flavescens Fabricius; Orthetrum sabino Drury; Coenagrion dyeri Fraser and Brachythemis contaminate Fabricius). Our results showed that microbiota densities and species in the dragonfly gut were rich and varied; these microbes showed complex interrelationships suggesting the host species had a major impact on gut community richness. The diversity of intestinal microbes may be related to the species and the habitats of dragonflies. Proteobacteria and Firmicutes were common in the four samples and represented core members of the dragonfly gut microbiota, accounting for approximately 99.00%. At the genus level, Serratia (83%), Lactococcus (41.5%), Hafnia (55.8%) and Lactococcus (71.7%) were dominant populations in the four groups (d1, h1_1, h1 and q1). Our results provide a basis for future research on gut bacteria functions in the dragonflies.
Introduction
Insects have successfully evolved to become the most diverse and abundant animal clade in terms of numbers of species, ecological habits and biomass [1]. Their evolutionary success has depended on their beneficial association with microorganisms, especially gut microorganisms [1-3]. Insect-microbes as mediators may be critical in the agriculture, medicine and ecology [4-6].
The diversity and versatility of insect-bacteria interactions represents enormous medical and agricultural potential in terms of modulation and control mechanism of insect populations [7]. Insects are responsible for massive agricultural losses and for pollination of many food crops, and microbes associated with pollinators and herbivores impact on crops, but they can also cause large agricultural losses [1].
Insects have evolved effective immune responses to combat pathogen infections, although insects, including honey bees, lack acquired immune responses [8-10]. Previous research has reported that gut microbes regulate host innate immunity and affects pathogen or parasite susceptibility [11-14]. In addition to beneficial effects, insect gut microbes may be harmful to their hosts [15,16]. Alterations in the gut microbiota of Drosophila, induced by the host’s deregulated immune genotype, may lead to host mortality [17]. Gut bacteria perturbations by antibiotics could increase susceptibilities to opportunistic bacterial pathogens and decrease survival of some insects, e.g. honey bees [18].
Insect gut microbiota has been analyzed using both culture-dependent and culture-independent methods [19-23]. Traditional culture methods often produce biased results since approximately 99% of all bacteria are uncultivable [24]. However, the development of meta-taxonomic analyses, based on high throughput sequencing of 16S rRNA, has greatly facilitated the profiling of microbial diversities in populations, when compared to traditional cultured-dependent and conventional molecular methods [25]. These comprehensive 16S rRNA sequence analysis projects provide dramatic insights into total bacterial diversity and metabolic activity of insect-associated microbial communities [26].
Dragonflies are important predators in both freshwater and terrestrial invertebrate foodwebs and are believed to be generalist predators that feed on a wide diversity of insects [27]. This dietary diversity should be associated with diverse gut microbial communities. The main objective of our study was to investigate bacterial diversity and composition of intestinal microbes and assess potential roles of gut bacteria in the digestion of four dragonfly species.
Materials and Methods
Ethics Statement
In our study four dragonfly species were investigated; Pantala flavescens Fabricius (q1); Orthetrum sabino Drury (h1); Brachythemis contaminate Fabricius (h1_1) and Coenagrion dyeri Fraser (d1). The flight abilities of the four species of dragonflies gradually diminished. At the same time, the four species are widely distributed throughout China, with no significant threats presently affecting these species (https://www.iucnredlist.org/search?taxonomies=100547 & search Type=species). No permits were required for catching dragonflies.
Sample collection
In July 2019, twenty dragonflies (five from each species) were caught using butterfly nets, at Jiangxi Normal University (28°66′87″N, 115°97′93″E), Nanchang city, Jiangxi province, PR China. P. flavescens and O. obsitina were caught in a meadow, while C. dyeri and B. contaminate adults were caught near a pond. Specimens were brought to the laboratory and were washed using 70% ethanol, followed by washing in distilled water [23]. Specimens were then stored at -20 until processing.
DNA extraction, amplification and high throughput Illumina sequencing
To determine gut bacterial composition, the gut was dissected and stored in a 1.5 ml tube for DNA extraction. Bacterial genomic DNA was extracted using the Wizard® Genomic DNA Purification Kit (Promega, Fitchburg, WI, USA) following manufacturer’s instructions. Extracted DNA was checked using 1% gel agarose electrophoresis, while the concentration and purity were determined using the NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). DNA from the four species was used as a temple to amplify the V3 - V4 region of the bacterial 16S rRNA gene using universal primers; 338F (ACTCCTACGGGAGGCAGCAG) and (GGACTACHVGGGTWTCTAAT). PCR reactions were performed on a GeneAmp® PCR 9700 System (ABI, Foster City, CA, USA) as previous described [28]. Once generated, PCR products were checked by 2% gel electrophoresis and extracted and purified using the DNA Gel Extraction Kit (Tiangen, Beijing, China) following manufacturer’s instructions. The purified DNA products were quantified and equalized using a TBS-380 Mini Fluorometer (Turner BioSystems, Sunnyvale CA, USA). The pooled products were submitted to Shanghai Majorbio Bio-Pharm Technology Co., Ltd (Shanghai, China) for 16S rRNA sequencing, using the Illumina Miseq PE300 platform (Illumina Corporation, San Diego, CA, USA).
Nucleotide sequences for all bacterial datasets were uploaded to NCBI (http://www.ncbi.nlm.nih.gov/) Short Read Archive, under the accession number PRJNA559000.
Data analysis
Raw Illumina read fastq files were de-multiplexed and quality filtered by Trimmomatic, and merged by FLASH [28]. We eliminated low quality sequences (quality scores lower than 20 over a 50 bp sliding window, primer mismatches lower than 2 and tags less than 50 bp). The length of overlap in merged sequences was over 10 bp and the wrong pair rate was low, 0.2. Usearch (version 7.1 http://drive5.com/uparse/) was applied to cluster the OTUs (Operational Taxonomic Units) at the identity threshold of 97%, and chimeric sequences were identified and removed by UCHIME [29,30]. Community alpha-diversity was analyzed using community diversity (Shannon and Simpson) and community richness (Chao and Ace) was analyzed using Mothur version v.1.30.1 [31].The Good’s coverage Shannon-Wiener curve and rarefaction curve were generated based on Mothur 1.30.1 software [31] with a 97% similarity. Phylogenetic beta diversity measures (hierarchical cluster tree and Non-MetricMulti Dimensional Scaling (NMDS), Venn diagrams, heatmap figures and rank-abundance curves were performed in vegan packages in R (version 3.1.2).
Phylogenetic affiliations from representative sequences were accessed using the Ribosomal Database Project(RDP) classifiers [32] against the Silva 16S rRNA database [33] with a confidence threshold of 70%. The community composition of each sample was analyzed for multiple taxa (phylum, class, order, family, genus and species). Bacterial similarities in different samples were calculated using hierarchical cluster analysis and un weighted UniFrac principal co-ordinates analysis (PcoA).
Results
Characteristics of sequencing data
From four samples, approximately 206,201 reads, with an average length of 449.28 bp were generated,using the Illumina Miseq sequence platform.Representative sequences for all OTUs are available (Short Read Archive, with the accession number PRJNA559000).Sequences were clustered into 115 OTUs and the Good’s coverage for observed OTUs was 99.99% ± 0.01% (Table 1).The alpha diversity for species richness (Chao), evenness (ACE) showed the highest diversity in the q1 sample, and the lowest diversity was in the h1 sample. Shannon’s diversity index and Simpson index showed the highest diversity in the h1-1 sample and the lowest diversity in the d1 sample (Table 1).
Table 1: MiSeq sequencing results and diversity estimates for each sample. * indicates 97% similarity.
| Sample ID |
Reads |
Diversity estimates* |
| OUT |
ACE |
Chao |
coverage |
Shannon |
Simpson |
| Coenagrion dyeri |
44572 |
50 |
53 |
54 |
0.999865 |
0.77 |
0.6954 |
| Orthetrum sabino |
44572 |
26 |
30 |
28 |
0.999888 |
1.24 |
0.3934 |
| Brachythemis contaminate |
44572 |
48 |
50 |
48 |
0.999955 |
1.91 |
0.2447 |
| Pantala flavescens |
44572 |
86 |
86 |
86 |
1 |
1.32 |
0.4339 |
The rarefaction curve indicated large variations in the total number of OTUs (Figure 1).

Figure 1 Rarefaction curves of OTUs in a 97% similarity boxplot for each sample.
The rarefaction curves all tended to approach saturation plateau,thereby indicating that the depth of sequencing was adequate for intestinal microbiota investigations.Among the four samples, the h1 sample had the lowest OTU density, followed by h1-1 and d1.
Taxonomic composition of samples from intestinal microbiota
Sequences that could not be classified into any known groups were unclassified.Bacterial OTUs were clustered into three phyla; Proteobacteria, Firmicutes and Bacteroidetes.Proteobacteria and Firmicutes were common to the four samples and represented core members of the dragonfly gut microbiota, accounting for 99.00% of total sequences (Figure 2).

Figure 2 Phylum distribution of gut samples.Other phyla were present but with less than < 0.01% abundance.
The d1 and h1 samples were dominated by Proteobacteria (93.28% and 98.89%,respectively).The q1 sample was dominated by Firmicutes (74.52%) and the h1-1 sample was dominated by Proteobacteria (50.52%) and Firmicutes (48.58%). Firmicutes, in the different samples showed high variability ranging from 74.52% to 1.11%. The final phyla, Bacteroidetes were also present, but with very low abundance (< 1%), except for q1 (approximately 1.1%).There were 13 identifiable bacterial genera which made up abundances of 98%, 100%, 100% and 98% in the four samples (Supplementary Figure 1). At the genus level, the d1 sample was dominated by Serratia (83%),Lactococcus (6.28%), Pantoea(3.41%),Enterobacter (3.24%), Proteus (1.85%) and other genera with less than 1% abundance.The h1-1 sample was dominated by Lactococcus(41.5%),Aeromonas (33.2%),Enterobacter(5.7%), Enterobaxteriaceae_unclassified (5.2%), Clostridium (4.9%), Pantoea (2.2%), Providencia (2.1%), unclassified genera (4.7%) and other genera with less than 1% abundance.The h1 sample was dominated by Hafnia (55.8%), Enterobacter (27.1%),Stenotrophomonas (7.90%),Pantoea (3.1%),Raoultella (2.7%),Serratia (1.9%) and other genera with less than1% abundance.The q1sample was dominated by Lactococcus (71.7%),Enterobacter (20%), Enterococcus (1.7%),Pantoea (1.5%),unclassified genera (4.3%) and other genera with less than 1% abundance (Supplementary Figure 1).
Difference and similarity of gut microbiota among four groups
Venn analysis was used to depict the shared or unique OTUs at the genus level. There was a total of 115 OTUs and 11 of them (10.4%) were shared genera (Figure 3).

Figure 3 Venn diagram showing bacterial genera detected in the four samples.Overlaps between samples are indicated by the arrangement of circles..
Q1 community contained more bacterial varieties (86 genera) than d1 (50), h1(26) and h1-1 (48). An overlap between the different communities was observed in Figure 3. The largest overlap was found between d1 and q1 libraries shared 44 of 92 OTUs; the q1 and h1_1 library shared 26 of the 108 (24%) OTUs, the q1 and h1 libraries with shared 22 of 90 (24%) OTUs. The h1 and h1_1 libraries shared 14 of the 81(17%) OTUs; d1 and h1 libraries shared 19 of 57 (33%) OTUs; d1 and h1_1 libraries shared 17 of 81 (21%) OTUs; h1, h1_1 and q1 libraries shared 13 of 94 (14%) OTUs, h1, h1_1 and d1 libraries shared 11 of the 85 (13%) OTUs, h1, q1 and d1 libraries shared 17 of the 94 (18%) OTUs, H1-1, q1 and d1 libraries shared 17 of 97 (18%) OTUs; h1, h1-1, q1 and d1 libraries shared 11 of 115 (9%) OTUs. D1 had 4 special OTUs, h1 had 1 special OTUs, h1-1 has 21 special OTUs and q1 had 30 special OTUs.
Based on genera relative abundance, genera with an average abundance of >1% in at least one sample were defined as predominant [34].There were three shared dominant genera (Enterobacter,Pantoea and Lactococcus) among the four samples OTUs, h1 had one special OTU, h1-1 had 21 special OTUs and q1 had 30 special OTUs (Figure 3).
PcoA was used to determine similarities in gut microbial communities in the four samples. Un-weighted UniFrac distance PcoA showed that the four samples were separate from each other, and that d1 was the most distinct (Supplementary Figure 2). Nonmetric multidimensional scaling (NMDS) based on Beta diversity also revealed distinct differences in species abundance across the four samples (Supplementary Figure 2).
Sample ID We also assessed similarities in gut microbiota at the phyla and genera level in all samples. Cluster analyses showed similar data (Figure 4).

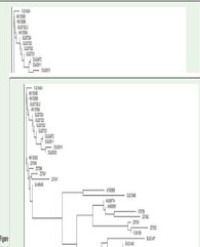
Figure 4 (A) Hierarchical cluster tree of samples based on intestinal microbiota composition. Bray-Cutis community similarity indices was used for community similarities and cluster analysis. (B) Major phyla distribution of gut microbiota among samples and the hierarchical cluster tree.
Q1 and h1 samples were clustered first, then h1-1 was clustered as one branch of (q1 + h1), and finally the d1 sample was clustered as one branch with groups of ((q1+ h1) and h1-1)) communities.
The network of co-occurrence of 16S rRNA gene function
Gut bacterial communities cannot be treated as single functional entities; the igraph R package was used to perform network analysis and visualization of OTUs. Gut microbial networks (Figure 5) consisted of 115 nodes (OTUs).

Figure 5 Co-occurrence network of 115 OTUs.
The OTUs were highly connected, revealing complex interrelationships between OTUs.
Discussion
Dragonflies are generalist predators in aquatic and associated terrestrial ecosystems [27], they consume a wide variety of insects and therefore harbor diverse gut microbial communities. Studies have investigated insect gut microbial communities to determine the prevalence of different microbes, their potential roles and their impact on host ecology and evolution [23]. Our results showed a large diversity of microorganisms displaying complex interrelationships in the dragonfly gut; the host species had a major impact on gut community richness. We observed substantial variation in the relative abundance of OTUs per species, which varied from 26 to 86 (Table 1). Similar patterns were also observed in three sympatric species of tsetse fly from Uganda [35]. In general, larger host species tend to harbor richer gut bacterial communities [23]. The richness of gut bacteria is associated with the habitat, ecological niche and predation ability of the host.
Pantala flavescens is a generalist species and consumes a diverse diet with excellent mobility; therefore P. flavescens had the largest OTU numbers [36-37]. C. dyeri had the smallest body but had the second largest number of OTUs, which may be related to its habitat. C. dyeri and B. contaminate shared similar habitats near water and preyed on small insects. O. sabino appeared to have lower OTU numbers when compared to C. dyeri and B. contaminate (Table 1), which may have been due to dietary specialization. Previous work has suggested that some dragonflies specialize on butterflies [38,39]. Recent analyses of sympatric dragonfly species have shown niche partitioning, with these dragonflies feeding on distinct species [40,41]. This dietary diversity may be associated with diverse gut microbial communities; however reports on dragonfly diets are rare. Nair and Agashe (2016) reported that OTU richness was strongly influenced by host species, the sampling month and interactions between the host sexes.
In our paper, dominant bacteria were similar in dragonfly gut bacterial communities. Proteobacteria and Firmicutes dominated and accounted for more than 97% in the gut community of dragonflies, which was also reported previously [23,42]. Shared genera included Serratia, Raoultella, Providencia, Pantoea, Lactococcus, Enterococcus, Enterobacter and Enterobacteriaceae_ unclassified. These shared genera may be related to basic functions of the host. Approximately 11 of 115 OTUs (10%) were shared across the four species. In contrast, a large portion of OTUs (approximately 56, 49% of the total) were rare and were found in single dragonfly species, e.g. Clostridium, Bacillus, Weissella, Chroococcidiopsis, Flavonifractor, Brochothrix. The existence of these exclusive genera may indicate specific functions within the host.
Hierarchical clustering, generated by gut communities, showed that evolutionary close dragonfly species had similar intestinal community compositions. These associations may result from facilitative interactions between bacteria from different genera, or they may indicate a common dietary (or environmental) source [23]. Data from un-weighted UniFrac distance PcoA and nonmetric multidimensional scaling also indicated similar associations.
In addition to interacting with the host, some bacterial species, isolated from damselfly feces, are perceived as disease causative agents, i.e. they responsible for urinary tract infections, pneumonia and septicemia in compromised hosts [43]. The fecal material of the damselfly can be used to monitor domestic antibiotic-resistant bacterial pollution associated with human health [43]. A metabolic disease of Libellula pulchella, caused by protozoan intestinal parasites, can result in the decline of mass specific fight muscle performance [44].
In summary, our results showed great diversity in gut microbiota from four dragonfly species. These gut microbiotas were highly connected, revealing complex interrelationship patterns. Gut community richness is often influenced by the host species; however functional mechanisms underlying gut microbiota diversity are poorly understood and require further research.
Acknowledgements
This research was supported by the National Natural Science Funds (No. 31960558) and the programs (GS201602 and 201622). We thank International Science Editing (http://www. International science editing.com) for editing this manuscript.
References
1. Engel P, Moran NA. The gut microbiota of insects - diversity in structure and function. FEMS Microbiol Rev.2013; 37: 699-735.
2. Kaufman, M. G. and Klug, M. J. The contribution of hindgut bacteria to dietary carbohydrate utilization by crickets (Orthoptera: Gryllidae). Comp Biochem Physiol. 1991; 98: 117–123.
3. Dillon R, Charnley K. Mutualism between the desert locust Schistocerca gregaria and its gut microbiota. Res Microbiol . 2002; 153: 503–509.
4. David Bignell, Paul Eggleton, Lina Nunes, Katherine L Thomas. Termites as mediators of carbon fluxes in tropical forests: budgets for carbon dioxide and methane emissions. Forests and Insects (Watt AD, Stork NE & Hunter MD, eds).1997; 109–134.
5. Fierer N, Strickland MS, Liptzin D, Bradford MA, Cleveland CC. Global patterns in belowground communities. Ecol Lett. 2009; 12: 1238 1249.
6. Fox-Dobbs K, Doak DF, Brody AK, Palmer TM. Termites create spatial structure and govern ecosystem function by affecting N2 fixation in an East African savanna. Ecology. 2010; 91: 1296-1307.
7. Shanchez-Contreras M, Vlisidou I. The Diversity of Insect-bacteria Interactions and its Applications for Disease ControlBiotechnol Genet Eng Rev. 2008; 25: 203-243.
8. Evans JD, Aronstein K, Chen YP, Hetru C, Imler JL, Jiang H, et al. Immune pathways and defence mechanisms in honey bees Apis mellifera. Insect Mol biol .2006; 15: 645-656.
9. Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu Rev Immunol. 2007; 25: 697-743.
10. Aggarwal K, Silverman N. Positive and negative regulation of the Drosophila immune response. BMB rep. 2008; 41: 267-277.
11. Buchon N, Broderick NA, Poidevin M, Pradervand S, Lemaitre B. Drosophila intestinal response to bacterial infection: activation of host defense and stem cell proliferation. Cell host & microbe. 2009; 5: 200-211.
12. Koch H, Schmid-Hempel P. Gut microbiota instead of host genotype drive the specificity in the interaction of a natural host-parasite system. Ecol Lett. 2012; 15: 1095-1103.
13. Cariveau DP, Elijah Powell J, Koch H, Winfree R, Moran NA. Variation in gut microbial communities and its association with pathogen infection in wild bumble bees (Bombus). ISME J. 2014; 8: 2369-2379.
14. Jiang Hong Li, Jay D. Evans, Wen Feng Li, Ya Zhou Zhao, Gloria DeGrandi-Hoffman, Shao Kang Huang, et al. New evidence showing that the destruction of gut bacteria by antibiotic treatment could increase the honey bee’s vulnerability to Nosema infection. PloS one. 2017; 12.
15. Broderick NA, Raffa KF, Handelsman J. Midgut bacteria required for Bacillus thuringiensis insecticidal activity. Proc Natl Acad Sci U S A. 2006; 103: 15196-15199.
16. Hurst GD, Jiggins FM. Male-killing bacteria in insects: mechanisms, incidence, and implications. Emerg Infect Dis. 2000; 6: 329–336.
17. Ryu JH, Kim SH, Lee HY, Bai JY, Nam YD, Bae JW, et al. Innate immune homeostasis by the homeobox gene caudal and commensal-gut mutualism in Drosophila. Science. 2008; 319: 777-782.
18. Raymann K, Shaffer Z, Moran NA. Antibiotic exposure perturbs the gut microbiota and elevates mortality in honeybees. PLoS Biol. 2017; 15: e2001861.
19. Wong CN, Ng P, Douglas AE. Low-diversity bacterial community in the gut of the fruitfly Drosophila melanogaster. Environ Microbiol. 2011; 13: 1889-1900.
20. Arias-Cordero E, Ping L, Reichwald K, Delb H, Platzer M, Boland W. Comparative evaluation of the gut microbiota associated with the below- and above-ground life stages (larvae and beetles) of the forest cockchafer, Melolontha hippocastani. PLoS One. 2012; 7: e51557.
21. Jones RT, Sanchez LG, Fierer N. A cross-taxon analysis of insect associated bacterial diversity. PLoS one. 2013; 8: e61218. Colman DR, Toolson EC, Takacs-Vesbach CD. Do diet and taxonomy influence insect gut bacterial communities? Mol Ecol. 2012; 21: 5124-5137.
22. Nair A, Agashe D. Host-specific spatial and temporal variation in culturable gut bacterial communities of dragonflies. Current science. 2016. 110: 1513-1523.
23. Rappé MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol. 2003; 57: 369 –394.
24. Ji-Hyun Yun, Seong Woon Roh, Tae Woong Whon, Mi-Ja Jung, Min-Soo Kim, Doo-Sang Park, et al. Insect Gut Bacterial Diversity Determined by Environmental Habitat, Diet, Developmental Stage, and Phylogeny of Host. Appl Environ Microb. 2014; 80: 5254 –5264.
25. Chen B, Teh BS, Sun C, Hu S, Lu X, Boland W. Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci Rep. 2016; 6: 29505.
26. Corbet, Philip S. Dragonflies: Behavior and Ecology of Odonata. Cornell University Press. 1999;
27. Zhang F, Ma T, Cui P, Tamadon A, He S, Huo C, et al. Diversity of the Gut Microbiota in Dihydrotestosterone-induced PCOS Rats and the Pharmacologic Effects of Diane-35, Probiotics, and Berberine. Front Microbiol.2019; 1 0: 175.
28. Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013; 79: 5112-5120.
29. Wei J, Guo X, Liu H, Chen Y, Wang W. The variation profile of intestinal microbiota in blunt snout bream (Megalobrama amblycephala) during feeding habit transition. BMC Microbiol. 2018; 18: 99.
30. Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 2011; 6: e27310.
31. Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2008; 37: D141-D145.
32. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013; 41: D590-D596.
33. Coburn B, Wang PW, Diaz Caballero J, Clark ST, Brahma V, Donaldson S, et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci Rep. 2015; 5: 10241.
34. Aksoy E, Telleria EL, Echodu R, Wu Y, Okedi LM, Weiss BL, et al. Analysis of multiple tsetse fly populations in Uganda reveals limited diversity and species-specific gut microbiota. Appl Environ Microbiol. 2014; 80: 4301–4312.
35. Daniel Troast , Frank Suhling, Hiroshi Jinguji, Göran Sahlén, Jessica Ware. Global Population Genetic Study of Pantala flavescens. Plos One. 2016; 11: e0148949.
36. Ling-zhen Cao, Kong-ming Wu .Genetic Diversity and Demographic History of Globe Skimmers (Odonata: Libellulidae) in China Based on Microsatellite and Mitochondrial DNA Markers. Sci Rep. 2019; 9: 8619.
37. Larsen TB. Butterflies as prey for Orthetrum austenti (Kirby) (Anisoptera: Libellulidae). Not Odonatol. 1981; 1: 130-133.
38. Owen DFOrthetrum austini Kirby, a specialist predator of butterflies (Anisoptera: Libellulidae), Not Odonato. . 1993; l 4: 34.
39. Michael L. May, Joel M. Baird. A comparison of foraging behavior in two “percher” dragonflies, Pachydiplax longipennis and Erythemis simplicicollis (Odonata: Libellulidae). J Insect Behav. 2002; 15: 765 778.
40. Khelifa R, Zebsa R, Moussaoui A, Kahalerras A, Bensouilah S, Mahdjoub H. Niche partitioning in three sympatric congeneric species of dragonfly, Orthetrum chrysostigma, O. coerulescens anceps, and O. nitidinerve: The importance of microhabitat. J Insect Sci. 2013; 13: 71.
41. Deb R, Nair A, Agashe D. Host dietary specialization and neutral assembly shape gut bacterial communities of wild dragonflies. PeerJ. 2019; 7: e8058.
42. Yamaguchi Y, Okubo T, Matsushita M, Wataji M, Iwasaki S, Hayasaka K. et al. Analysis of adult damselfly fecal material aids in the estimation of antibiotic resistant Enterobacterales contamination of the local environment. Peer J. 2018; 6: e5755
43. Schilder RJ, Marden JH. Metabolic syndrome and obesity in an insect. Proc Natl Acad Sci U S A. 2006; 103: 18805-18809.
Citation
Cao L, Bu X (2020) The Composition and Interaction of the Gut Microbiota in Four Species of Wild Dragonflies. Ann Appl Microbiol Biotech nol J 4: 7. doi: https://dx.doi.org/10.36876/apmbj607280